BY ANISH KOKA
Reanalysis of a trial used to approve a commonly used injectable cholesterol-lowering drug confirms the original analysis by accident.
The open-data movement seeks to liberate the massive amount of data generated in running clinical trials from the grasp of the academic medical-pharmaceutical industrial complex that mostly runs the most important trials responsible for bringing novel therapeutics to market.
There are only a few elite academic trialist groups capable of running large trials and there’s ample reason to be suspicious about the nexus that has developed between academia and the pharmaceutical companies that shower them with cash to hopefully get a positive study result and pay off the pharmaceutical research investment manifold. The FDA is the major regulator of the whole process, but the expertise required for regulation means that the FDA is frequently comprised of ex-pharma employees or ex-academics.
The voluminous data generated in trials is usually owned by the academic group doing the research and is frequently guarded heavily from outsiders who may seek to probe it. Many, of course, believe that the data should be made public for independent third-party review.
Discovering discrepancies in a major published trial from the pharma-academic complex would be a boost to those seeking to force trial data to be public, and that is exactly what a group of investigators attempted to do with a major cholesterol-lowering trial published in 2017.
But first, some background.
Cholesterol-lowering is big business.
The global market for the most commonly used class of drugs used to improve cholesterol levels is $14.3 billion dollars. Highly potent statins are now generic, accessible to most Americans for ~$4/month, and when tolerated, very effective at reducing a sticky molecule that floats around in the bloodstream called low-density lipoprotein (LDL).
LDL floats around in the bloodstream and is thought to incrementally accumulate on blood vessels in the body, ultimately increasing the risk of heart attacks and strokes. There is some controversy about whether LDL is the best metric to follow with regards to cardiovascular risk, but there are a wealth of randomized control trials of a variety of therapeutics that reduce LDL and also reduce the risk of heart attacks and strokes (the LDL hypothesis).
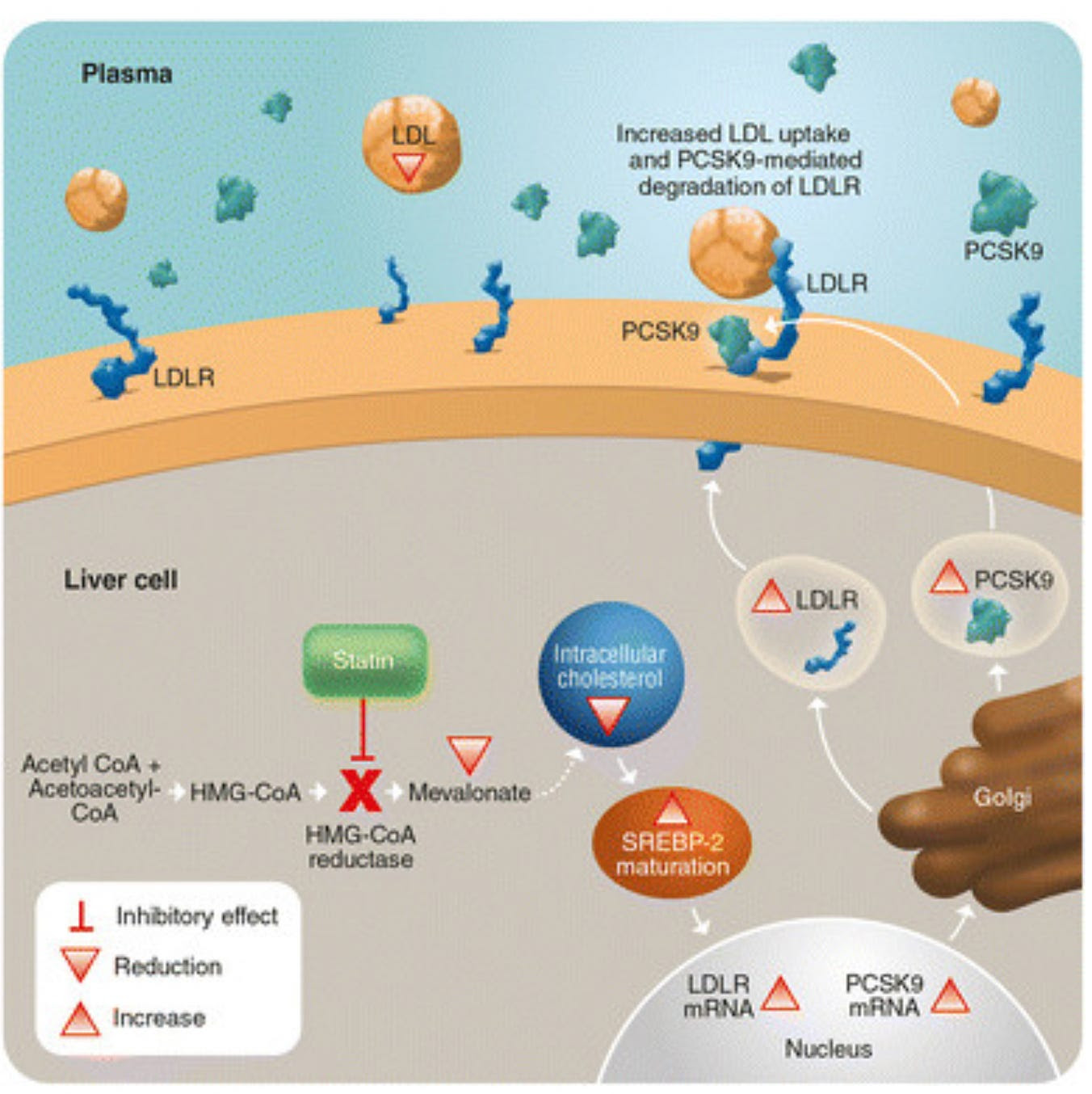
Statins inhibit the intracellular liver enzyme HMG-CoA reductase, which then results in up-regulation of LDL receptors on the surface of the liver cell. These receptors “catch” LDL particles that are floating by and incorporate them into the liver cell leading to less LDL in the bloodstream to cause vascular problems.
But while statins are believed to have been a big part of the reduction in cardiovascular morbidity seen in the last three decades, people still have heart attacks and strokes.
The obvious question when dealing with this residual cardiovascular risk is whether driving LDL concentrations down even further would significantly lower or even abolish heart attacks and strokes.
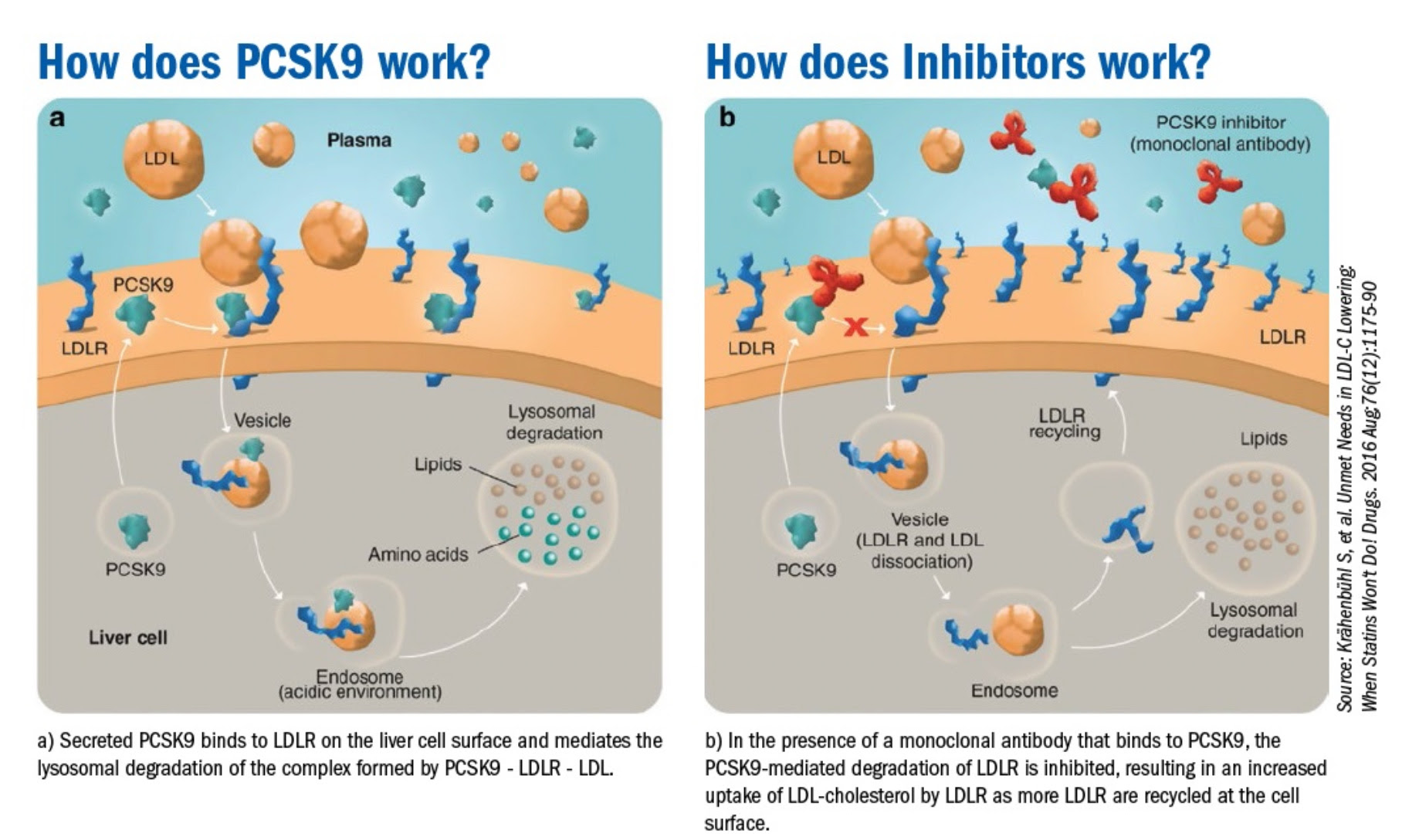
The effort to suppress LDL even further lead to the development of an entirely new class of lipid lowering agents that boosted the concentration of LDL receptors on the liver cell surface. Scientists noted an intracellular protein called PCSK9 combined with LDL and the LDL receptor lead to degradation of the entire complex in the cell’s incinerator otherwise known as the lysosome. A monoclonal antibody developed to bind the PCSK9 protein (a PCSK9 inhibitor) that prevented formation of the PCSK9-LDL receptor-LDL complex, prevented the LDL receptor from being degraded in the lysosome, and resulted in recycling of the LDL receptor to the cell surface. Use of a PCSK9 inhibitor in addition to a statin results in much higher concentration of LDL receptors on the cell surface than with a statin alone, which results in profound lowering of LDL well beyond what was seen with statins alone.
Proving benefit, and ensuring safety of this novel class of drugs with potential wide applicability requires large randomized clinical trials, and the FOURIER trial was just that vehicle. Trials of this size are complex and can’t be done by just anyone, which is why the famed TIMI group was tasked with the job. The TIMI (Thrombolysis in Myocardial Infarction) Study Group is a Division of Cardiovascular Medicine at the esteemed Brigham and Women’s Hospital and Harvard Medical School.
Those of us hoping that super low LDL levels would abolish cardiovascular disease were pretty disappointed with the results of the trial published in 2017. While the drug destroyed LDL levels in blood compared to the control arm, it didn’t come anywhere close to abolishing cardiovascular disease.
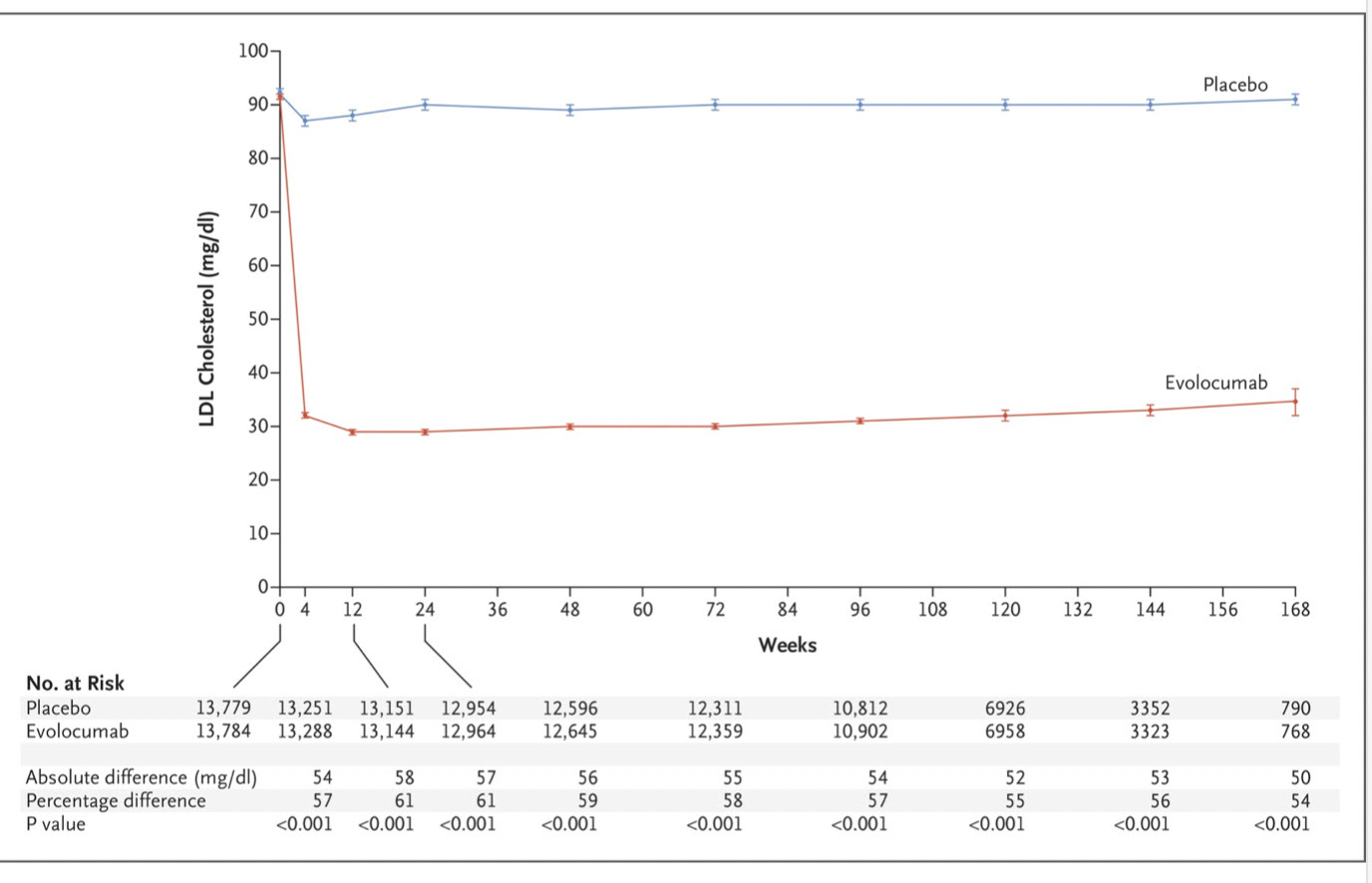
It did, however, reduce a composite cardiovascular endpoint by a statistically significant 1.5% which greatly impressed some clinical trialists.
But the primary endpoint here was a composite endpoint of cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization. The positive results described were driven almost entirely by fewer heart attacks and fewer revascularization procedures in the evolocumab arm. Importantly, there was no difference in cardiovascular death or death from any cause.
As a brief aside: The PCSK9 inhibitor class of drugs were approved by the FDA because of trials like FOURIER, and the drug hit the US market with a list price of $14,000 / year. Insurance companies balked at those prices and put up road blocks to patient access by way of onerous prior authorization processes. The makers of the 2 PCSK9 inhibitors eventually buckled and slashed list prices of the drug by 60% in 2019 in an effort to expand use of the drug.
The FOURIER results piqued the curiosity of another group of investigators who were suspicious about certain irregularities they noticed. The Restoring Invisible and Abandoned Trials (RIAT) initiative is an international effort to tackle bias in research reporting whose goal is to provide more accurate information to patients and other healthcare decision makers. This group noticed the numerically higher (but statistically insignificant) number of cardiovascular deaths and all cause deaths in the evolocumab arm in the FOURIER trial. They also noted that most cardiovascular deaths (n=372/491; 75.8%) were classified as ‘other cardiovascular death’, not as myocardial infarctions or congestive heart failure, which typically predominate among cardiovascular deaths.
To the RIAT group, this was grounds for a re-analysis based on the Clinical Study Report (CSR) that provides more abundant and detailed evidence than had been published. The CSR is a technical document prepared by the manufacturer and submitted to regulators as part of the approval package for drug evaluation. It contains information about the trial’s protocol, amendments, inclusion and exclusion criteria, outcome definitions and measurement, efficacy and safety results and statistical analysis plan. The main FOURIER CSR is over 25,000 pages.
The RIAT analysis, published in the BMJ Open found that for 360/870 deaths (41.4%), the cause of death adjudicated by the FOURIER clinical events committee differed from that declared by the local clinical investigator. After readjudication, deaths of cardiac origin were numerically higher (though still statistically insignificant) in the evolocumab group than in the placebo group in the FOURIER trial, suggesting possible cardiac harm. Based on this, the RIAT authors advised “clinicians should be sceptical about prescribing evolocumab for patients with established atherosclerotic cardiovascular disease.”
The gravity of the charges made can’t be understated. The RIAT group essentially was calling out the prestigious TIMI investigators, alleging incompetence.. or worse. They were also calling into question the FDA oversight of the trial that was supposed to be doing the job of verifying the data submitted. It made for a neat story for those suspicious of the pharmaceutical-medical complex and the FDA regulators frequently supplied from academia and industry.
The TIMI group was not shy in their response.
Writing in a rapid response comment to the RIAT analysis, the TIMI group noted they had a lot more information that was available to the RIAT investigators to adjudicate adverse events.
The article by Erviti et al. is fundamentally flawed, using incomplete data to reach incorrect conclusions. In FOURIER, 2 event adjudication followed a rigorous, pre-specified, blinded process by the TIMI Clinical Events Committee (CEC). The occurrence of potential cardiovascular events of interest triggered collection of a full dossier containing all relevant and available source documents, including hospital notes, laboratory, ECG and imaging data, procedure reports, resuscitation or code summaries, death certificates, and autopsy reports. Each dossier was independently evaluated by 2 experienced, board-certified cardiologists (cardiovascular events) or neurologists (cerebrovascular events), blinded to treatment allocation.
The TIMI group also noted the FDA audited the adjudication process, and questioned the training the RIAT investigators had to adjudicate deaths.
The adjudication charter was approved by the FDA before the trial commenced. At the end of the trial the FDA audited the adjudication process and results and had no findings of concern.
In contrast, the authors’ work was post hoc and relied only on one document: the CSR narrative, which was generated predominantly based on limited information provided by the site upon learning of the event and not intended for the purpose of formal event adjudication. It is unclear what training and expertise, if any, those classifying events for this paper had.
Even more devastating was the TIMI group’s examination of two examples of mistaken death adjudication the RIAT group had forwarded in their BMJ Open analysis. It turns out the RIAT death adjudication process consisted primarily of re-affirming the local investigators initial cause of death. The TIMI group pointed out that the job of their clinical events committee was to investigate each death using local source documents and reclassify deaths when appropriate.
In the first case, the authors state the patient “clearly” had an MI, which was “neglected” by the FOURIER CEC. In reality, the source documents show this patient died in his sleep, and thus according to the FDA definitions, the FOURIER (TIMI) CEC properly adjudicated this as sudden cardiac death. In the second case, the authors state the patient died of an MI that was “misadjudicated” by the FOURIER CEC as a non-cardiovascular death. In reality, the source documents show this patient slipped in his kitchen, struck his head, was admitted to the ED with head trauma and died, which would not be considered a CV death.
It would appear to the objective observer that the RIAT group may have had good intentions, but had no idea what they were doing. The other curiosity here is the editorial freedom given the RIAT authors to publish a warning to physicians about prescribing these drugs based on a signal of harm that was essentially fabricated. Given how careful authors must be in their wording in manuscripts when presenting positive results of drug/device trials, it would appear there’s a fair degree of editorial bias that allows the publication of salacious claims about a pharmaceutical company and the group tapped to run their trials with much less evidence. At the very least, a discriminating editor would have vetted the allegations with the TIMI group before publishing a document that suggested clinicians be cautious of prescribing a drug because of a signal of harm.
But beyond this particular trial, and the controversy surrounding the poorly done reanalysis there are important questions regarding the responsibility trialists have for transparency of data. The reanalysis failed in this case because the RIAT authors did not have access to the same data the primary investigators had. It should be particularly galling that there is no easy mechanism for the public to access primary source documents even in taxpayer-funded trials. The current approach gives a virtual monopoly to a few large trialist groups like the TIMI group and makes the FDA the only oversight body. At a time when trust in the pharmacy-academic industrial complex and the public institutions is at an all time low, it’s understandable there’s a hunger for independent analysis and oversight.
This would require a significant infrastructure investment , and clearly badly done analyses performed by amateurs would only worsen the signal-to-noise ratio and confuse the public.
The Open Data movement stumbled badly here, but in doing so provides much food for thought.
Anish Koka is a cardiologist. Follow him on twitter @anish_koka
Categories: Medical Practice